Last reviewed: March 2026
Contents
MDM Templates
Acetaminophen Overdose
Patient presents after intentional acetaminophen ingestion. Well appearing on arrival. No abdominal pain, no jaundice, no altered mental status.
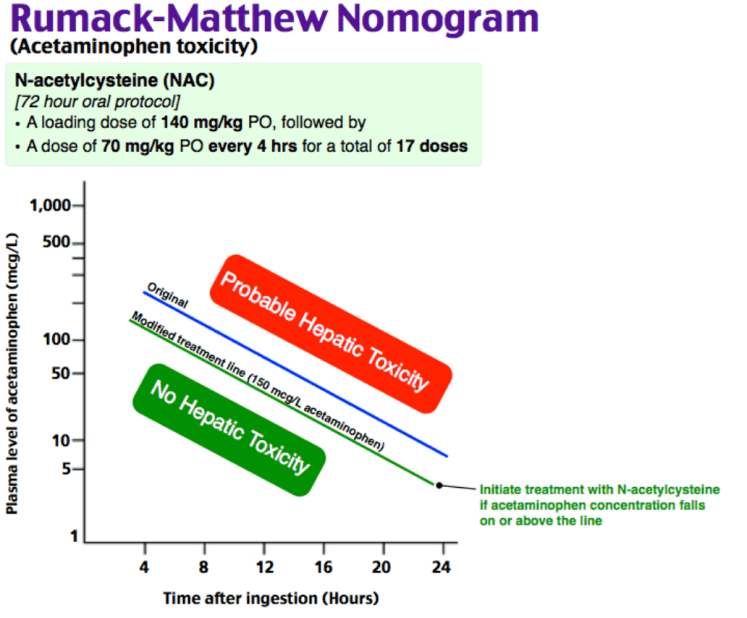
Acetaminophen toxicity is the leading cause of acute liver failure in the US. Patients are often asymptomatic in the first 24 hours despite potentially lethal ingestion. The 4-hour APAP level plotted on the Rumack-Matthew nomogram determines treatment. Must also rule out co-ingestion — acetaminophen and salicylate levels are mandatory in all intentional ingestions regardless of stated substance.[1]
Plan: 4-hour acetaminophen level (or immediate level if >4 hours since ingestion). If level above treatment line on Rumack-Matthew nomogram, initiate NAC. LFTs and INR as baseline. If LFTs elevated, start NAC regardless of APAP level. Psychiatry consulted for intentional ingestion. Admit for NAC completion and psychiatric evaluation.
Salicylate Toxicity
Patient presents after salicylate ingestion with tinnitus, tachypnea, nausea, and diaphoresis. Labs notable for mixed respiratory alkalosis and metabolic acidosis.
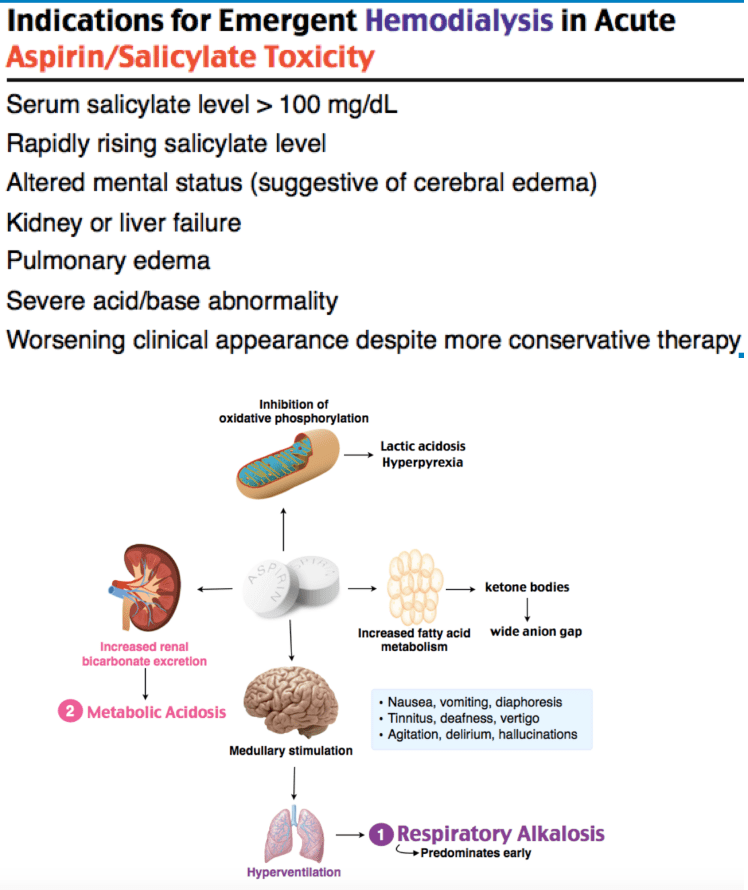
Salicylate toxicity causes direct stimulation of the respiratory center (respiratory alkalosis), uncoupling of oxidative phosphorylation (metabolic acidosis), and altered mental status from CNS toxicity. The mixed acid-base picture is characteristic. Salicylates also cause non-cardiogenic pulmonary edema and cerebral edema in severe cases. Must differentiate from sepsis, DKA, and other causes of anion gap acidosis.[2]
Plan: Serial salicylate levels (levels can rise for hours after ingestion, especially with enteric-coated or bezoar formation). Urinary alkalinization with sodium bicarbonate (3 amps NaHCO3 in 1L D5W, goal urine pH 7.5-8.0). Monitor potassium aggressively — hypokalemia prevents urinary alkalinization. Hemodialysis if salicylate level >100 mg/dL acute (or >60 mg/dL chronic), altered mental status, pulmonary edema, renal failure, or clinical deterioration despite alkalinization. Psychiatry consulted for intentional ingestion. Admit to ICU for severe toxicity.
TCA Toxicity
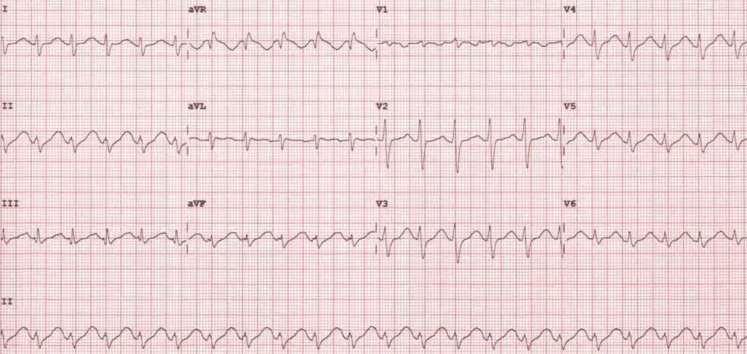
Patient presents after intentional ingestion of tricyclic antidepressant with altered mental status, tachycardia, and widened QRS on ECG. Anticholinergic toxidrome present — mydriasis, dry mucous membranes, urinary retention, decreased bowel sounds.
TCA toxicity causes a combination of anticholinergic effects, sodium channel blockade (wide QRS, dysrhythmias), and alpha-adrenergic blockade (hypotension). The sodium channel blockade is the lethal mechanism — it causes wide-complex dysrhythmias and cardiovascular collapse. Seizures are common and worsen acidosis, which increases the ionized fraction of TCA available to block sodium channels, creating a vicious cycle.[3]
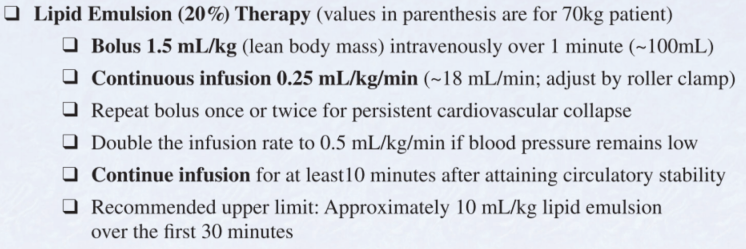
Plan: Sodium bicarbonate boluses (50 mEq IV, repeat q2 min) until QRS narrows below 100 ms, then bicarbonate infusion (150 mEq in 1L D5W). Magnesium 2g IV empirically. Benzodiazepines for seizures. Norepinephrine for hypotension. Avoid class IA/IC antiarrhythmics. Intubation with hyperventilation (RR 24-30) if needed to maintain alkalosis. Admit to ICU with continuous cardiac monitoring.
Carbon Monoxide Poisoning
Patient presents with headache, nausea, dizziness, and confusion after enclosed-space exposure. Carboxyhemoglobin level elevated. Other household members symptomatic.
Carbon monoxide binds hemoglobin with 200-250x the affinity of oxygen, displacing oxygen and shifting the oxyhemoglobin dissociation curve to the left, impairing oxygen delivery to tissues. Standard pulse oximetry is unreliable — it cannot distinguish carboxyhemoglobin from oxyhemoglobin and will read falsely normal. Diagnosis requires co-oximetry on ABG.[4]
Plan: High-flow oxygen via non-rebreather (half-life of COHb drops from 5 hours on room air to 90 minutes on 100% O2). Continuous cardiac monitoring. Troponin if chest pain or ECG changes. Hyperbaric oxygen therapy if loss of consciousness, syncope, neurologic deficits, carboxyhemoglobin >25%, or pregnancy with COHb >15%. Identify and remove the CO source. Evaluate all co-exposed individuals.
Cyanide Toxicity
Patient presents with altered mental status, lactic acidosis, and hemodynamic instability after fire exposure involving synthetic materials, or after industrial chemical exposure. Lactic acidosis out of proportion to clinical picture.
Cyanide uncouples the electron transport chain by inhibiting cytochrome oxidase, forcing anaerobic metabolism and producing profound lactic acidosis. Tissues cannot extract oxygen, so venous oxygen saturation is paradoxically high (“arterialization” of venous blood). Diagnosis is clinical — serum cyanide levels are not available in real-time. Must consider concurrent carbon monoxide poisoning in fire victims.[4]
Plan: Hydroxocobalamin (Cyanokit) 5g IV over 15 minutes — binds cyanide directly to form cyanocobalamin (vitamin B12), which is renally excreted. Treat empirically based on clinical suspicion — do not wait for levels. Concurrent high-flow oxygen for possible CO co-exposure. Sodium thiosulfate is an alternative if hydroxocobalamin is unavailable. Admit to ICU.
Methemoglobinemia
Patient presents with cyanosis, headache, and oxygen saturations in the mid-80s that do not improve with supplemental oxygen. Exposure to oxidizing agent identified (dapsone, benzocaine, nitrites).
Methemoglobin contains oxidized iron (Fe3+) that cannot bind oxygen, reducing oxygen-carrying capacity. Pulse oximetry is unreliable — it reads the absorption spectrum of methemoglobin and trends toward 85% regardless of true saturation. Diagnosis requires co-oximetry on ABG showing elevated methemoglobin fraction. Notably, there is no lactic acidosis in methemoglobinemia (unlike cyanide) because tissues can still extract the oxygen that is delivered.[4]
Plan: Methylene blue 1-2 mg/kg IV over 5 minutes for methemoglobin >25% or any symptomatic patient (beyond headache). Methylene blue acts as an electron carrier to reduce Fe3+ back to Fe2+. Supplemental oxygen. Remove offending agent. Repeat methylene blue dose if no improvement in 30 minutes (max 7 mg/kg total). Methylene blue is contraindicated in G6PD deficiency (causes hemolytic anemia) — use ascorbic acid instead. Admit for monitoring.
Clinical Education
Acetaminophen Pearls
Toxic single acute dose in adults is 7.5-10 grams (approximately 15-20 extra-strength tablets). The daily maximum recommended dose is 3-4 grams. Many patients are unaware of acetaminophen content in combination products (Vicodin, Percocet, NyQuil, Excedrin).[1]
NAC is the antidote and is nearly 100% effective if given within 8 hours of ingestion. IV NAC protocol: 150 mg/kg over 60 minutes loading dose, then 50 mg/kg over 4 hours, then 100 mg/kg over 16 hours. Anaphylactoid reactions (flushing, urticaria, bronchospasm) can occur with the loading dose — slow the infusion and give diphenhydramine, do not discontinue NAC.
NAC is safe in pregnancy. Acetaminophen overdose in pregnancy is treated the same as non-pregnant patients. Do not withhold NAC due to pregnancy.
Chronic/repeated supratherapeutic ingestion is a different entity than acute overdose. The Rumack-Matthew nomogram does not apply. Obtain APAP level, LFTs, and INR. Treat with NAC if LFTs are elevated or APAP level is detectable, regardless of nomogram.
AST > ALT pattern is characteristic of acetaminophen hepatotoxicity (AST rises first and higher). Transaminases can reach 10,000+ IU/L. Similar pattern is seen with amatoxin poisoning.
Salicylate Pearls
Salicylate toxicity produces a characteristic mixed acid-base disturbance: respiratory alkalosis (from direct respiratory center stimulation) AND metabolic acidosis (from uncoupled oxidative phosphorylation and accumulation of organic acids). Early in toxicity, the respiratory alkalosis may predominate. As toxicity worsens, the metabolic acidosis takes over.[2]
Potassium must be repleted aggressively during urinary alkalinization. The kidney preferentially retains potassium over hydrogen ions — if the patient is hypokalemic, the kidney will dump protons instead of retaining them, and the urine will remain acidic no matter how much bicarbonate you give. Keep K >4.0 mEq/L.
Intubation in salicylate toxicity is extremely dangerous. The patient’s compensatory tachypnea (RR 30-40) is maintaining a respiratory alkalosis that keeps the salicylate ionized and out of the CNS. If you intubate and ventilate at a normal rate (RR 14-16), the resulting drop in pH will drive salicylate into the brain, causing rapid deterioration. If intubation is necessary, match the patient’s pre-intubation minute ventilation (high RR, high TV).
Hemodialysis indications: Salicylate level >100 mg/dL (acute) or >60 mg/dL (chronic), altered mental status, pulmonary edema, renal failure, or failure to improve with alkalinization.
TCA Pearls
The ECG is the most important prognostic tool in TCA toxicity. QRS >100 ms predicts seizures. QRS >160 ms predicts ventricular dysrhythmias. A terminal R wave in aVR >3 mm is highly suggestive of sodium channel blockade (though this finding may not appear until after bicarbonate administration).[3]
Sodium bicarbonate is the antidote. It works by two mechanisms: (1) increasing serum sodium to competitively bind sodium channels, and (2) alkalinizing the blood to increase protein-bound (non-toxic) TCA fraction. Goal pH 7.50-7.55. Use QRS width as the titration endpoint (goal <100 ms).
If seizing, consider pyridoxine (B6) — TCA-related seizures are GABAergic, and pyridoxine is the antidote for isoniazid (INH) seizures which have a similar mechanism. In an unknown polypharmacy overdose, giving pyridoxine 5g IV is reasonable.
Avoid: Class IA/IC antiarrhythmics (worsen sodium channel blockade), physostigmine (may cause asystole in TCA toxicity), flumazenil (can precipitate seizures). Activated charcoal is generally not recommended due to aspiration risk with rapidly declining mental status.

Intralipid emulsion therapy (lipid rescue) can be considered for refractory cardiovascular collapse from TCA toxicity. 1.5 mL/kg 20% lipid emulsion IV bolus, then 0.25 mL/kg/min infusion.
Carbon Monoxide Pearls
Pulse oximetry is falsely reassuring in CO poisoning. Standard pulse oximeters measure only two wavelengths and cannot distinguish oxyhemoglobin from carboxyhemoglobin. A patient with COHb of 30% may show SpO2 of 99%. Always use co-oximetry (ABG) for diagnosis.[4]
Sources of CO exposure: House fires, gas-powered generators, furnaces, water heaters, car exhaust in enclosed spaces, charcoal grills used indoors. Consider CO poisoning when multiple household members present with similar symptoms (headache, nausea, confusion).
Delayed neuropsychiatric syndrome (DNS) occurs in 10-30% of CO-poisoned patients, developing days to weeks after apparent recovery. Symptoms include cognitive deficits, personality changes, parkinsonism, and dementia. Hyperbaric oxygen therapy may reduce (but does not eliminate) DNS risk.
Hyperbaric oxygen indications: Loss of consciousness, syncope, focal neurologic deficits, seizures, cardiac ischemia, COHb >25%, pregnancy with COHb >15%, persistent symptoms despite normobaric oxygen. Transfer to the nearest HBO facility if criteria met.
Serotonin Syndrome vs NMS
These two syndromes are commonly confused but have critical distinguishing features:
| Feature | Serotonin Syndrome | Neuroleptic Malignant Syndrome |
| Onset | Hours (rapid, within 24h of exposure) | Days to weeks after starting/increasing antipsychotic |
| Key exam finding | Clonus and hyperreflexia | Lead-pipe rigidity |
| Pupils | Mydriasis | Normal |
| Bowel sounds | Hyperactive | Decreased |
| Skin | Diaphoretic | Diaphoretic |
| Treatment | Benzodiazepines, cyproheptadine 12 mg PO/NG | Benzodiazepines, dantrolene, bromocriptine |
| Resolution | 24-72 hours after removing agent | Days to weeks |
Serotonin syndrome: Caused by excess serotonergic activity, most commonly from drug combinations (SSRI + MAOI, SSRI + tramadol, SSRI + linezolid). May also cause SIADH with hyponatremia and QTc prolongation. Monitor for delayed seizures up to 13 hours after ingestion. MAOI overdoses should be monitored 24 hours due to risk of delayed deterioration.
NMS: Caused by dopamine blockade, typically from antipsychotics started or increased within the prior 2 weeks. Complications include rhabdomyolysis, renal failure, hepatic failure, and DIC. CK is markedly elevated. Consult poison control regarding dantrolene and bromocriptine.
Disposition
Discharge: Acetaminophen with non-toxic 4-hour level and normal LFTs (still requires psychiatric evaluation before discharge if intentional). Carbon monoxide with mild symptoms resolved on NRB and COHb normalizing. Methemoglobinemia resolved after methylene blue with methemoglobin <5%.
Admit (floor/telemetry): Acetaminophen requiring NAC protocol completion. Carbon monoxide requiring prolonged oxygen therapy. Mild salicylate toxicity responding to alkalinization. Methemoglobinemia requiring repeat dosing or monitoring.
Admit (ICU): TCA toxicity (any QRS widening or hemodynamic instability). Severe salicylate toxicity (AMS, pulmonary edema, HD indicated). Cyanide poisoning. Serotonin syndrome or NMS with hyperthermia or hemodynamic instability. Any ingestion with refractory acidosis, hemodynamic compromise, or airway concerns.
References
- Heard KJ. Acetylcysteine for acetaminophen poisoning. N Engl J Med. 2008;359(3):285-292. PubMed
- Palmer BF, Clegg DJ. Salicylate toxicity. N Engl J Med. 2020;382(26):2544-2555. PubMed
- Liebelt EL. Targeted management strategies for cardiovascular toxicity from tricyclic antidepressant overdose: the pivotal role for alkalinization and sodium loading. Pediatr Emerg Care. 1998;14(4):293-298. PubMed
- Weaver LK. Carbon monoxide poisoning. N Engl J Med. 2009;360(12):1217-1225. PubMed
